



Research
Ion Conduction in Solids and Liquids
The mobility of small ions (H+, Li+) in condensed matter is governed by perpetual bond breaking and formation processes. Inspired by its crucial relevance for batteries and fuel cells, we analyze the atomic dynamics on picosecond timescales in several systems with first principles molecular dynamics simulations.
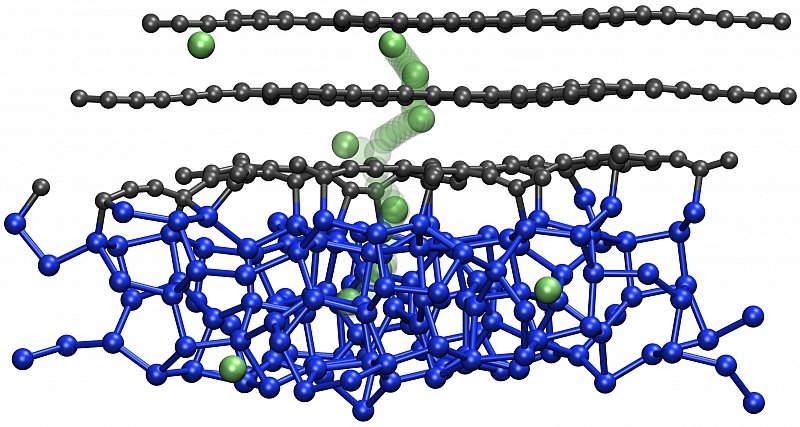
Li+ dynamics in silicon is important for the next-generation high-capacity batteries using amorphous silicon as anode storage material for lithium. Our focus here is at the quantitative modeling of geometric pathways and the kinetics of Li+ dynamics in a variety of lithium-silicide compounds, as well as Si/C interfaces. The aim is to understand diffusion mechanisms and energy barriers to develop advanced models for multiscale simulations of Li diffusivity in Si anode materials. [10.1021/acs.jpcc.2c01555 ]
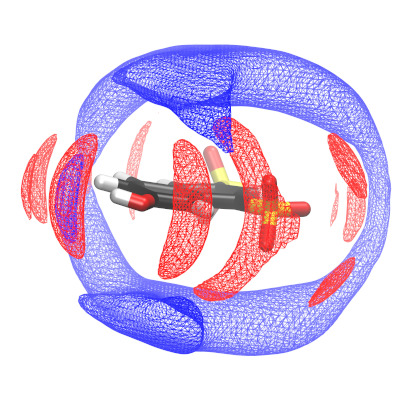
H+ dynamics in complex liquids and phosphonic acids derivatives is relevant in both fuel cell membrane materials as well as biochemical systems. In a joint experimental/computational project, we investigate the proton dynamics in different amphoteric solvent mixtures using molecular dynamics simulations. We analyze the local solvation environment of a photoacid which serves as a controlled proton injector, and derive the probability distributions of donor-acceptor pairs to identify preferential proton pathways. In a related project, we study the proton mobility in a series of phosphate and sulfate salts (Cs H2PO4, Cs HSO4) and its derivatives, with the focus of describing large-scale and long-time dynamics (up to micrometer and microseconds) at a quasi-atomistic level.
Kinetic/Lattice Monte Carlo
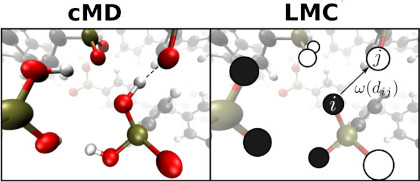
We develop a combined molecular dynamics/Monte Carlo simulation technique, which allows the efficient simulation of bond breaking/formation processes (such as ion dynamics) up to microsecond and micrometer time- and length scales. The particular feature is that simulated data is channeled from quantum chemical calculations and molecular dynamics simulations to a more coarse-grained description combining methodic elements of kinetic and lattice Monte Carlo techniques. This combination allows the incorporation of picosecond atomistic dynamics into large-scale effective diffusion simulations, e.g. for the accurate modeling of ion dynamics around mesoscopic structural defects. [10.1039/C7CP05632J , 10.1021/acs.jpcc.6b05822 ]
Explicit Configurational Entropy in Molecular Simulations
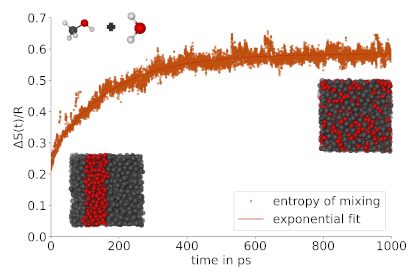
We have developed a novel method to compute the configurational entropy of mixing at the quantitative level. The method allows the direct observation of mixing/demixing processess in molecular simulation, relying exclusively on the atomic coordinates (i.e. the trajectory) and is applicable to isolated as well as periodic simulations. The approach is able to describe the formation of local structural heterogeneities (cluster formation, micro-phase separation) as well as global phase separation phenomena at a high accuracy. Our tool offers an exciting opportunity to apply fundamental thermodynamics to advanced simulations. [10.1021/acs.jpclett.4c02819 ]
Polyphilicity

The concept of philicity describes the relative affinity of a molecular component to interact with several other components in a mixture. This project aims at understanding this qualitative concept from elementary interaction values of functional groups which are available quantitatively at the simulation level. We modify systematically the strength of individual intermolecular interactions in force field molecular dynamics simulations of binary mixtures, which allows to determine which molecular property is responsible for which ``type'' of philicity in the mixture. The aim is eventually to better understand the role of molecular groups in the miscibility of complex polyphilic systems.
Cellulose
![Ionic liquid [EMIm][OAc], DMSO and water around a cellulose molecule](/im/1733737583_1576_0.png)
Cellulose, a sustainable biopolymer, is challenging to process due to poor solubility in conventional solvents. Its dissolution in ionic liquids like [EMIm][OAc] is studied using force field molecular dynamics simulations, with a focus on hydrogen bonding networks. The effects of co-solvents like DMSO on enhancing dissolution and anti-solvents such as water on inducing precipitation are explored.[10.1039/D2CP05636D , 10.1007/s10570-024-05854-4 ]
Back-mapping from Coarse-grained to Atomistic Structures
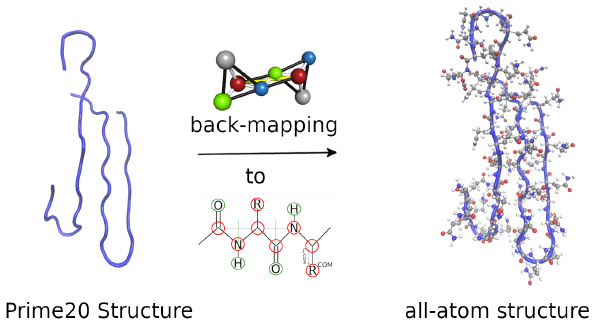
The concentional coarse-graining approach replaces functional groups (e.g. short alkyl chains, amino/amide groups) with effective particles in view of achieving a more efficient phase space sampling (via molecular dynamics or Monte Carlo simulation techniques). We investigate the conformational results of such coarse-graining schemes by reconstructing the atomistic structure and subsequently benchmarking its local structural stability. This tool allows a validation of the quality of structural results obtained from coarse-graining approaches. [doi.org/10.1002/cphc.202300521 , doi.org/10.1002/cphc.202400592 ]
Battery Materials (Jr. Group Partovi)
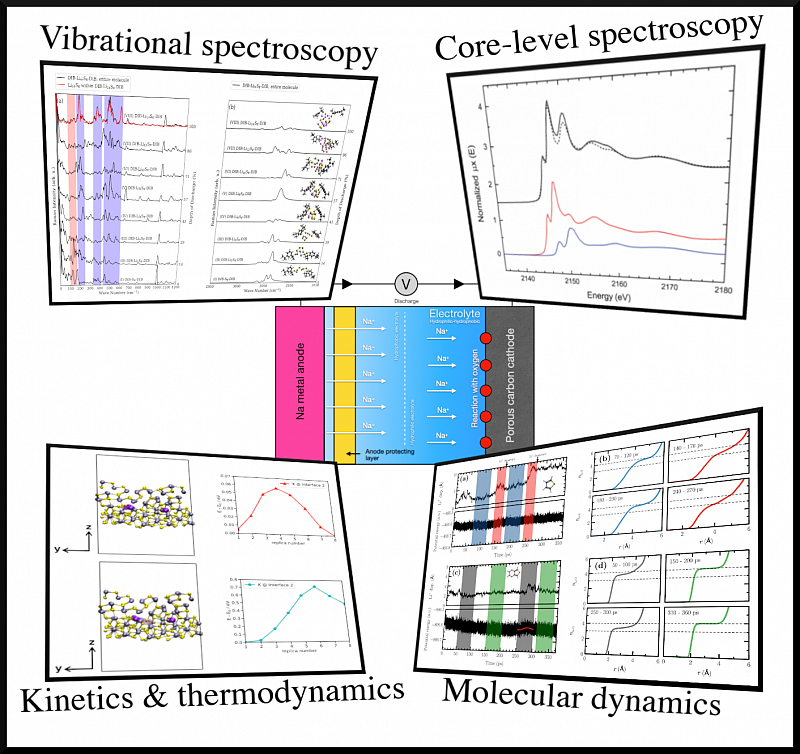
Global energy demands require more efficient energy storage and conversion systems. As conventional batteries approach their energy density limits, alternative technologies are essential. Using classical and quantum methods alongside spectroscopy simulations, we aim to characterize the atomistic processes in lithium and sodium batteries. We also investigate the (electro)catalytic properties of platinum-group metals for energy conversion [https://doi.org/10.1002/cphc.202400681 , https://doi.org/10.1021/acs.chemmater.7b05105 ].