



Forschung
Ionen-Mobilität in Feststoffen und Flüssigkeiten
Die Mobilität kleiner Ionen (H+, Li+) in kondensierter Materie wird durch ständige Bindungsbruch- und -bildungsprozesse bestimmt. Aufgrund ihrer entscheidenden Bedeutung für Batterien und Brennstoffzellen analysieren wir die atomare Dynamik auf Pikosekunden-Zeitskalen in verschiedenen Systemen mit Simulationen der Molekulardynamik nach ersten Prinzipien.
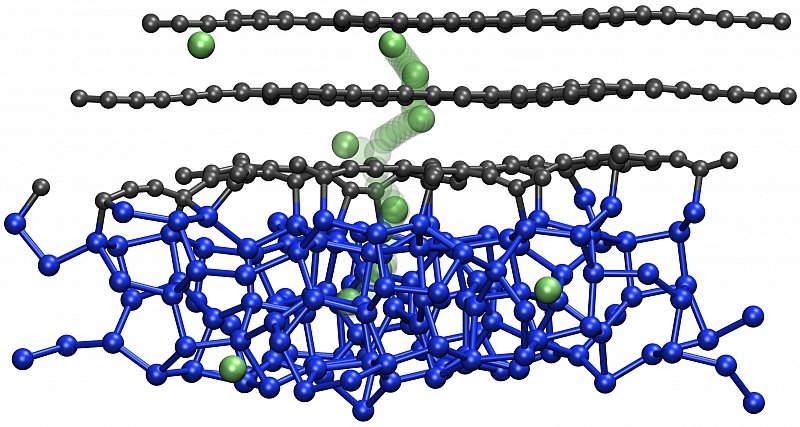
Die Li+-Dynamik in Silizium ist wichtig für die nächste Generation von Hochleistungsbatterien, die amorphes Silizium als Anodenspeichermaterial für Lithium verwenden. Unser Schwerpunkt liegt hier auf der quantitativen Modellierung der geometrischen Pfade und der Kinetik der Li+-Dynamik in einer Vielzahl von Lithium-Silizid-Verbindungen sowie an Si/C-Grenzflächen. Ziel ist es, Diffusionsmechanismen und Energiebarrieren zu verstehen, um fortschrittliche Modelle für Multiskalensimulationen der Li-Diffusionsfähigkeit in Si-Anodenmaterialien zu entwickeln. [10.1021/acs.jpcc.2c01555 ]
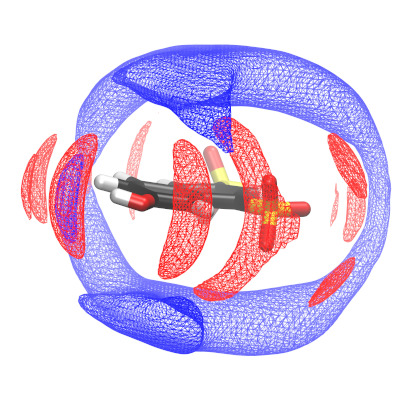
Die H+-Dynamik in komplexen Flüssigkeiten und Phosphorsäurederivaten ist sowohl für Brennstoffzellenmembranen als auch für biochemische Systeme von Bedeutung. In einem gemeinsamen experimentellen und rechnergestützten Projekt untersuchen wir die Protonendynamik in verschiedenen amphoteren Lösungsmittelgemischen mit Hilfe von Molekulardynamiksimulationen. Wir analysieren die lokale Solvatationsumgebung einer Photosäure, die als kontrollierter Protoneninjektor dient, und leiten die Wahrscheinlichkeitsverteilungen von Donor-Akzeptor-Paaren ab, um bevorzugte Protonenpfade zu identifizieren. In einem verwandten Projekt untersuchen wir die Protonenmobilität in einer Reihe von Phosphat- und Sulfatsalzen (Cs H2PO4, Cs HSO4) und ihren Derivaten, wobei der Schwerpunkt auf der Beschreibung der großräumigen und langzeitlichen Dynamik (bis zu Mikrometer und Mikrosekunden) auf quasi-atomarer Ebene liegt.
Kinetic/Lattice Monte Carlo
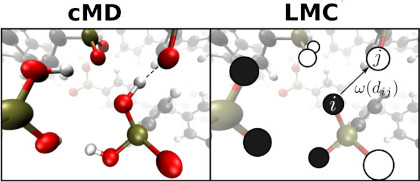
Wir entwickeln eine kombinierte Molekulardynamik-/Monte-Carlo-Simulationstechnik, die die effiziente Simulation von Bindungsbruch-/Bildungsprozessen (wie z. B. Ionendynamik) bis zu Zeit- und Längenskalen im Mikrosekunden- und Mikrometerbereich ermöglicht. Die Besonderheit besteht darin, dass die simulierten Daten von quantenchemischen Berechnungen und Molekulardynamiksimulationen zu einer grobkörnigeren Beschreibung geleitet werden, die methodische Elemente von kinetischen und Gitter-Monte-Carlo-Techniken kombiniert. Diese Kombination ermöglicht die Einbeziehung der atomistischen Dynamik im Pikosekundenbereich in großmaßstäbliche effektive Diffusionssimulationen, z. B. für die genaue Modellierung der Ionendynamik um mesoskopische Strukturdefekte. [https://doi.org/10.1039/C7CP05632J , https://doi.org/10.1021/acs.jpcc.6b05822 ]
Explizite Konfigurationsentropie in Molekularsimulationen
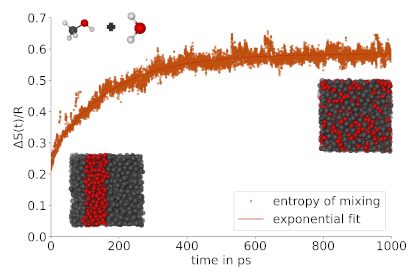
Wir haben eine neuartige Methode entwickelt, um die Konfigurationsentropie der Vermischung auf quantitativer Ebene zu berechnen. Die Methode ermöglicht die direkte Beobachtung von Mischungs-/Entmischungsprozessen in der Molekularsimulation, wobei sie sich ausschließlich auf die Atomkoordinaten (d.h. die Trajektorie) stützt und sowohl auf isolierte als auch auf periodische Simulationen anwendbar ist. Der Ansatz ist in der Lage, sowohl die Bildung lokaler struktureller Heterogenitäten (Clusterbildung, Mikrophasentrennung) als auch globale Phasentrennungsphänomene mit hoher Genauigkeit zu beschreiben. Unser Werkzeug bietet eine aufregende Möglichkeit, grundlegende Thermodynamik auf fortgeschrittene Simulationen anzuwenden. [10.1021/acs.jpclett.4c02819 ]
Polyphilizität

Das Konzept der Philizität beschreibt die relative Affinität einer molekularen Komponente zur Wechselwirkung mit mehreren anderen Komponenten in einem Gemisch. Dieses Projekt zielt darauf ab, dieses qualitative Konzept anhand von elementaren Wechselwirkungswerten funktioneller Gruppen zu verstehen, die auf der Simulationsebene quantitativ verfügbar sind. Wir modifizieren systematisch die Stärke einzelner intermolekularer Wechselwirkungen in Kraftfeld-Molekulardynamiksimulationen von binären Gemischen, was es ermöglicht, zu bestimmen, welche molekulare Eigenschaft für welchen ``Typ'' von Philizität im Gemisch verantwortlich ist. Ziel ist es, die Rolle der molekularen Gruppen bei der Mischbarkeit komplexer polyphiler Systeme besser zu verstehen.
Cellulose
![Cellulosemolekül umgeben von der ionischen Flüssigkeit [EMIm][OAc], DMSO und Wasser](/im/1733737583_1576_0.png)
Cellulose, ein nachhaltiges Biopolymer, ist aufgrund seiner schlechten Löslichkeit in herkömmlichen Lösungsmitteln schwer zu verarbeiten. Die Löslichkeit in ionischen Flüssigkeiten wie [EMIm][OAc] wird mit Hilfe von Kraftfeld-Molekulardynamik-Simulationen untersucht, wobei der Schwerpunkt auf Wasserstoffbrücken-Netzwerken liegt. Die Auswirkungen von Co-Solvents wie DMSO auf die Verbesserung der Löslichkeit und von Anti-Solvents wie Wasser auf die Ausfällung von Cellulose werden untersucht. [10.1039/D2CP05636D , 10.1007/s10570-024-05854-4 ]
Back-mapping von grobkörnigen zu atomistischen Strukturen
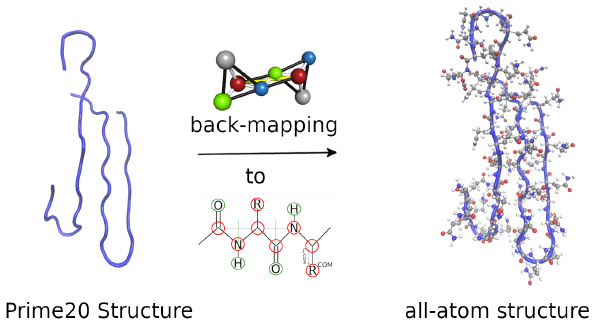
Bei diesem Ansatz werden funktionelle Gruppen (z. B. kurze Alkylketten, Amino-/Amidgruppen) durch effektive Teilchen ersetzt, um eine effizientere Phasenraumabtastung (mittels Molekulardynamik oder Monte-Carlo-Simulationstechniken) zu erreichen. Wir untersuchen die Konformationsergebnisse solcher Grobkornschemata durch Rekonstruktion der atomistischen Struktur und anschließendes Benchmarking ihrer lokalen strukturellen Stabilität. Dieses Instrument ermöglicht eine Validierung der Qualität der strukturellen Ergebnisse, die mit Grobkornverfahren erzielt werden. [doi.org/10.1002/cphc.202300521, doi.org/10.1002/cphc.202400592 ]
Battery Materials (Jr. Group Partovi)
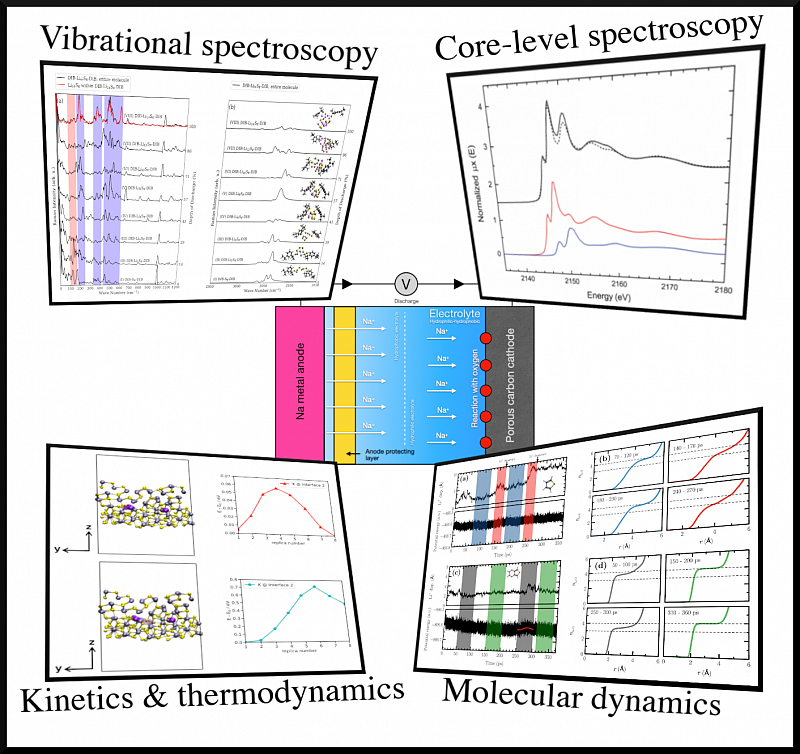
Der weltweite Energiebedarf erfordert effizientere Systeme zur Energiespeicherung und -umwandlung. Da herkömmliche Batterien an die Grenzen ihrer Energiedichte stoßen, sind alternative Technologien unerlässlich. Mit klassischen und Quantenmethoden sowie Spektroskopiesimulationen wollen wir die atomistischen Prozesse in Lithium- und Natriumbatterien charakterisieren. Außerdem untersuchen wir die (elektro)katalytischen Eigenschaften von Platingruppenmetallen für die Energieumwandlung [https://doi.org/10.1002/cphc.202400681 , https://doi.org/10.1021/acs.chemmater.7b05105 ].